
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |
- 낙농식품미생물학
- 환경생명화학
- 미생물학실험
- NGS
- consensus sequence
- gene cloning
- 내재면역
- 미생물학
- MHC
- 적응면역
- 기능성식품과면역학
- Expasy
- Average Nucleotide Identity
- 단백질구조
- EC number
- 식물영양학
- 생물정보학
- 유전공학의기초
- 원핵생물
- 자가항원
- BLaST
- Bcell
- Swissprot
- Eztaxon
- phylogenetic tree
- 미생물의생장
- 체액성면역
- 면역학정리
- 면역학
- primer design
- Today
- Total
Self Love is the Best Love
2. Phylogenetic tree and Average Nucleotide Identity (ANI) 본문
1. Purpose
Phylogenetic tree는 생물이 진화의 결과 여러 종이나 분류군 사이에서 나타나는 신체적이거나 유전적 특징의 유사성과 차이를 바탕으로 evolutionary relationship을tree diagram으로 나타낸 것이다[1]. 다양한 생물 종에 대해 진화적 관계를 알 수 있다.
Phylogenetic tree를 구성하기 위해, 분자 진화에 대한 통계적 분석을 수행하는 Molecular Evolutionary Genetics Analysis (MEGA)이라는 컴퓨터 소프트웨어를 사용한다[2]. 또한 종의 재분류를 방지하기 위해 Average Nucleotide Identity (ANI)를 이용해 두 유전체의 코딩된 부분 사이의 nucleotide수준의 Genomic Similarity을 측정한다.
이 실험에서는 MEGA 프로그램을 통해 주어진 미생물들의 Phylogenetic tree를 그리고, ANI calulator로 임의의 두 미생물의 Genomic Similarity을 측정하려고 한다.
2. Materials & Methods
2.1 Phylogenetic tree그리기
미생물의 염기 서열을 포함하는 text파일의 확장자를 .fasta로 새로 저장한다.(Pic2) FASTA format은base pair 혹은 amino acids이 단일 문자 코드를 사용하여 표현되는 nucleotide sequences 혹은 peptide sequences을 나타내는 텍스트 기반 형식이다. Text 파일에서 “>”는sequence의 시작을 의미한다. Fasta file은 공백 인식이 되지 않기 때문에 공백 대신 - 혹은 _을 사용해 띄어쓰기를 표시한다. (Pic1) 확장자를 .fasta로 저장한다.
 Pic1. 미생물의 염기 서열을 포함하는 text |
 |
MEGA 프로그램을 실행시킨다. 상단의 Align에서 Edit/build alignment를 클릭한다. Create a new alignment. DNA alignment를building한다. Pic2.에서 저장한 fasta파일을 MEGA로drag and drop. (Pic3)
 Pic3. MEGA로 불러온 FASTA file |
전체 서열을 선택하고 상단의W아이콘을 클릭한다. Align DNA. 설정을 바꾸지 않고 OK한다. 전체 서열에서 상동성이 있는 부분은 sequence위에 * mark가 생긴다. (Pic4)
 Pic4. * 표시된 sequences. |
Sequence 앞과 뒤에 * mark가 없는 부분을 삭제한다. (Pic5)
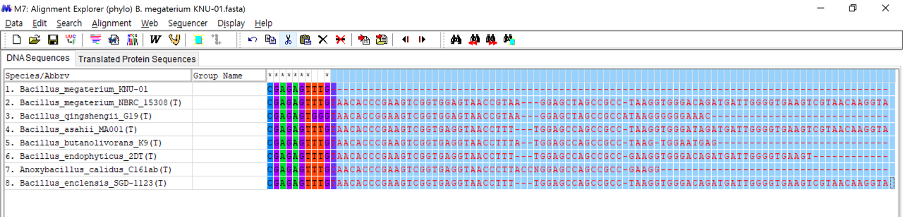 Pic5. * mark가 없는 서열 |
이후 상단의 Data에서 Export alignment. MEGA format으로 저장한다. 이를 다시 MEGA프로그램으로 불러온다. 상단의 Phylogeny에서 Construct/Text Neighbor-Joining Tree를 한다. (Pic6) 현재 파일을 사용할 것이냐는 안내 문구가 뜬다. Yes. (Pic6)
 Pic6. MEGA로 불러옴 |
Option Summary창에서 parameter를 설정한다. Phylogeny Test의Test of Phylogeny를Bootstrap method로 하고 No. of Bootstrap Replication을1000으로 설정한다. Substitution Model의Substitutions Type을Nucleotide로 하고 Model/Method는Juke-Cantor model로 설정한다. (Pic7) Compute.
 Pic7. Set parameter |
2.2. Genomic Similarity 측정
비교하고자 하는 whole genome sequence를text파일에서 FASTA파일로 따로 저장한다. EzTaxon site를 통해 ANI Calculator를 들어간다. 저장했던 FASTA파일을 업로드한다. (Pic8)
 Pic8. 비교하고자 하는 두 미생물의 sequence FASTA파일 업로드 |
3. Results & Discussions
MEGA프로그램으로 그린 미생물들의 Phylogenetic tree는 다음과 같다. (Pic9)
 Pic9. Phylogenetic tree |
ANI Calculator로 계산한 Bacillus asahii와Bacillus butanolivorans의ANI value는97%이다. 즉, 두 미생물은 97%의 Genomic Similarity를 가지고 있다. (Pic10)
 Pic10. Result of ANI calculator |
- OrthoANI value(%) : ANI percentage
- Genome A length (bp) : genome size of A
- Genome B length (bp) : genome size of B
- Average aligned length (bp) : aligned sequence length from genome A and B
- Genome A coverage (%) : percentage of covered sequence when genome A was put as query
- Genome B coverage (%) : percentage of covered sequence when genome B was put as query
4. References
[1] Pulves외, 이광웅 외 역, 생명 생물의 과학, 2006, 교보문고, ISBN 89-7085-516-5, 제3부 진화의 과정, 제23장 계통의 재구성과 활용
[2] Kumar,s., K. Tamura및M. Nei (1993) MEGA : Molecular Evolutionary Genetics Analysis. Ver. 1.0, The Pennsylvania State University, University Park, PA.
'20211 > 생물정보학' 카테고리의 다른 글
| 4. Protein identification using Swissprot (Expasy) (0) | 2021.08.16 |
|---|---|
| EC number (0) | 2021.08.16 |
| 3. EC number and protein blast (0) | 2021.08.16 |
| 1. Sanger sequencing and Eztaxon (Identification): sequencing merge & running Eztaxon (0) | 2021.08.13 |
| Next Generation Sequencing(NGS)의 종류와 원리, 활용 분야 (0) | 2021.08.13 |

